.png)
We introduce triplet envelope functions as a new concept for machine learning interatomic potentials (MLIPs) that improve training and inference efficiency and stability. By pruning local atomic neighborhoods through physically inspired geometric screening, we achieve significant computational gains without sacrificing accuracy. Specifically, the triplet envelope function doubles training and inference speed, triples memory efficiency, and ensures energy conservation—a critical factor for reliable molecular dynamics simulations.
MLIPs have revolutionized materials science by offering the accuracy of density functional theory (DFT) energy and forces with linear computational scaling. Of particular importance was the discovery that universal interatomic potential could be trained from expansive datasets to accurately simulate materials across the periodic table. To accomplish this, general-purpose machine learning interatomic potentials (MLIPs) had to adopt larger radial cutoffs (typically 6 Å). This was so that even low density materials could be accurately modeled. However, this came with increased costs for more dense materials. Because the density of materials varies significantly, the number of neighbors for high density materials can be almost 10x greater than smaller neighbors. This asymmetry causes specific challenges for memory management and cost, requiring complex error handling of out-of-memory events.
Previous attempts to address this asymmetry have used K-Nearest Neighbor (KNN) sparsification to force all nodes to have a maximum number of edges in a graph. KNN sparsification shows negligible impact on accuracy while solving memory and cost concerns. This shows that a significant portion of neighbors provide essentially no value. Unfortunately, KNN sparsification breaks energy conservation, leading to incorrect observables from molecular dynamics simulations.
To address the memory, cost, and energy conservation, we introduce a new concept of higher-order envelope functions that extend the two-body radial envelope function commonly used in MLIPs. We showcase a specific type of higher order envelope function, a triplet envelope function that achieves the same benefits of KNN sparsification without losing energy conservation. In particular, we utilize a triplet envelope function based on the screening term in the classical modified embedded atom method (MEAM) interatomic potential.
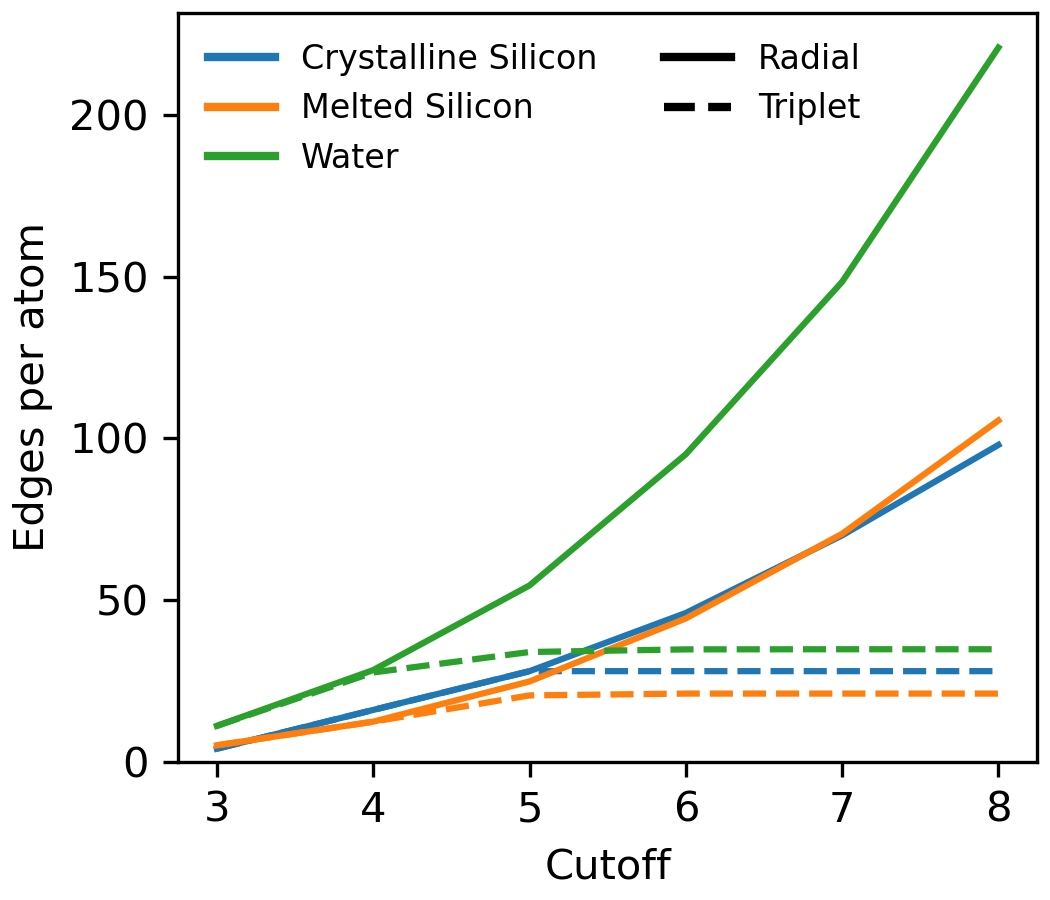
The triplet envelope function reduces the number of neighbors each atom has in its local neighborhood. Figure 1 depicts this neighborhood sparsification for three different systems: crystalline silicon, molten silicon, and water. In these environments, the radial environment can have more than 3x more neighbors in the neighborhood defined by the radial as compared to the triplet envelope function.
Figure 1: Comparison of neighborhoods defined by radial and triplet envelope functions for a radial cutoff of 6Å. The top two rows depict the local neighborhood of a central atom (blue), where the top row is colored by chemical identity and the middle row by the triplet screening parameter. The bottom row depicts the local environments after pruning with the triplet envelope function.
The introduction of the triplet envelope function has multifaceted benefits across training and inference efficiency, memory optimization, simulation stability, and energy conservation.
A full description of the methods and results can be found in our paper: https://arxiv.org/abs/2602.02228.
The triplet envelope function offers a principled method to address the asymmetric memory and cost of different environments seen when training and using general purpose MLIPs on systems that span solids, liquids, and gases. Furthermore, it paves the way for using larger cutoff radii without a prohibitive performance penalty to better model complex, open structures like metal-organic frameworks (MOFs). Finally, beyond immediate performance, ablation studies comparing radial and triplet envelope functions provide a deep insight into MLIP design: a significant portion of neighbors provide essentially no additional information. The triplet envelope function therefore provides a geometric inductive bias that allows models to focus on the most impactful physical interactions, paving the way for the next generation of stable and high-performance materials simulations.