
We use PHIN-OS to simulate the Horiuti–Polanyi mechanism for acetylene hydrogenation, a widely used process for purifying industrial hydrocarbon streams. The case study validates a reproducible digital workflow that can subsequently be used to rapidly develop novel, cost-effective catalysts.
Catalysts make industrial processes economical by reducing the energy and time it takes to transform matter between states. Hydrogen catalysts in particular play a central role in the oil and gas industry by hydrogenating hydrocarbons. In particular, the Horiuti–Polanyi mechanism involves the decomposition of hydrogen on a catalyst for use in hydrogenating hydrocarbons in a step-by-step process. This reaction is critical for purifying ethylene streams by removing trace contaminants like acetylene that can contaminate downstream polymerization catalysts.
Palladium is one of the most widely used Horiuti–Polanyi catalysts due to its balance of binding hydrogen molecules while not overbinding them so that they can be released. Furthermore, palladium has a barrierless hydrogen decomposition pathway providing a steady stream of protons for hydrogenating acetylene. However, the performance of palladium comes at a price. Palladium is a platinum group catalyst with a high cost. Finding a PGM-free alternative therefore remains an active area of research.
Here we use PHIN-OS to create a simulation workflow for simulating the Horiuti–Polanyi process for a palladium catalyst. We start with investigating the barrierless transition of hydrogen on palladium and then a mixture of hydrogen with acetylene on palladium to simulate the hydrogenation process. Validating the ability for PHIN to simulate the Horiuti–Polanyi process serves as a precursor to designing novel PGM-free catalysts by creating a reproducible baseline.
Atomic scale simulations are well suited for studying chemical reactions due to their ability to mechanistically isolate the bond rearrangements happening at the atomic scale. Here we use a combination of energy minimization and NVT simulations to study the hydrogenation of acetylene on palladium.
The first simulation workflow focuses on the barrierless decomposition of hydrogen on palladium to confirm the accuracy of the interatomic potential. After confirming the accuracy of the model, the second simulation workflow combines minimization and NVT simulations to predict the hydrogenation of acetylene on palladium through the Horiuti–Polanyi.
The barrierless decomposition of hydrogen on palladium is validated by adsorbing a single hydrogen molecule on a palladium surface. The hydrogen molecule is placed in the “on-top” position and allowed to relax using a conjugate gradient relaxation algorithm.
The initial position and the subsequent relaxation is seen in the video in Figure 1. The hydrogen decomposes into the palladium hollow sites as reported from experimental observations and previous DFT investigations. The video shows that the location of the hydrogen molecule is insensitive to the initial guess as it is able to diffuse on the surface laterally before decomposing.

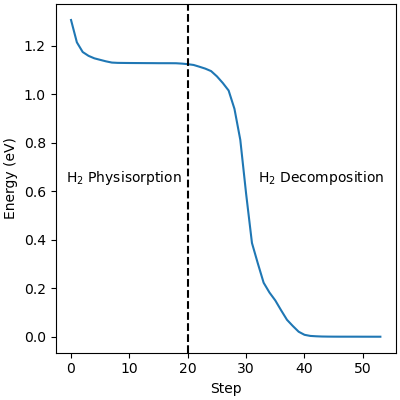
The atomistic reaction is supplemented with the reaction thermodynamics. The minimization energy trace in Figure 2 confirms the presence of two different regimes. The first regime physisorbs hydrogen to the surface and, due to the barrierless nature of hydrogen decomposition, progresses immediately into the second hydrogen decomposition pathway. The minimization continues on a nearly flat plateau until it finds the H2 decomposition pathway with each proton settling in adjacent hollow sites. The reaction thermodynamics are exothermic, releasing just over an eV of energy. This is in good agreement with experimental values, confirming that the interatomic potentials are appropriate for simulating catalytic chemistry.
Now that we’ve confirmed the accuracy of the interatomic potentials on hydrogen decomposition, we move to the more complicated acetylene hydrogenation reactions, which combine hydrogen decomposition with acetylene to simulate the Horiuti–Polanyi mechanism. The simulation of the hydrogenation reaction requires a more complicated process and is best visualized with PHIN-OS. Figure 3 shows the 8 steps process for the digital simulation of the Horiuti-Polanyi mechanism. After constructing the palladium surface, a mixture of hydrogen gas and acetylene are added to model a low-pressure low-temperature heterogeneous catalyst reaction. After creating the model system, we subsequently minimize the energy which prepares the atomic structure for simulated decomposition.
After finding the minimum energy palladium surface, four hydrogen and one acetylene molecule are added to the simulation. Similar results to minimizing the isolated hydrogen atoms are found but with more complexity due to the multiple hydrogen molecules and acetylene. The structural information in Figure 4 shows the physisorption of acetylene and hydrogen followed by the decomposition of two hydrogen molecules. The energy trace in Figure 3b shows the multiple different intermediate states that the system traverses through until a metastable state is accessed.
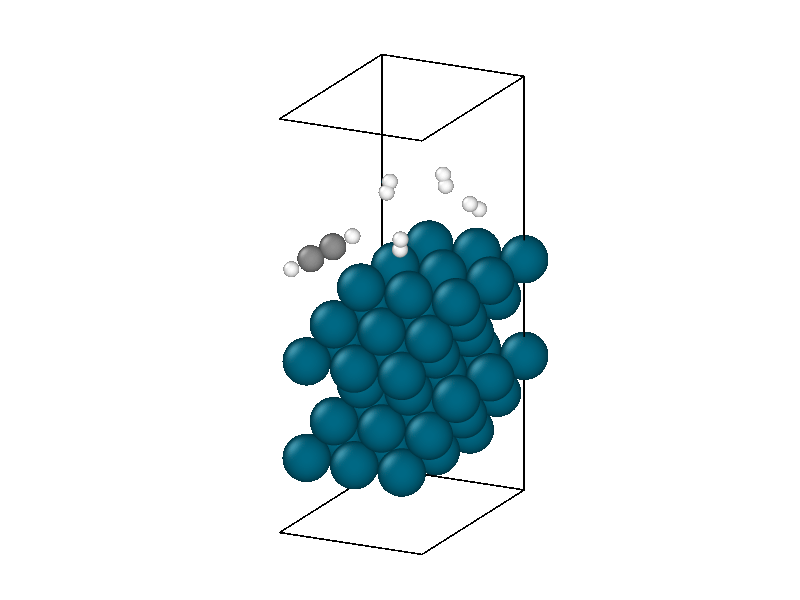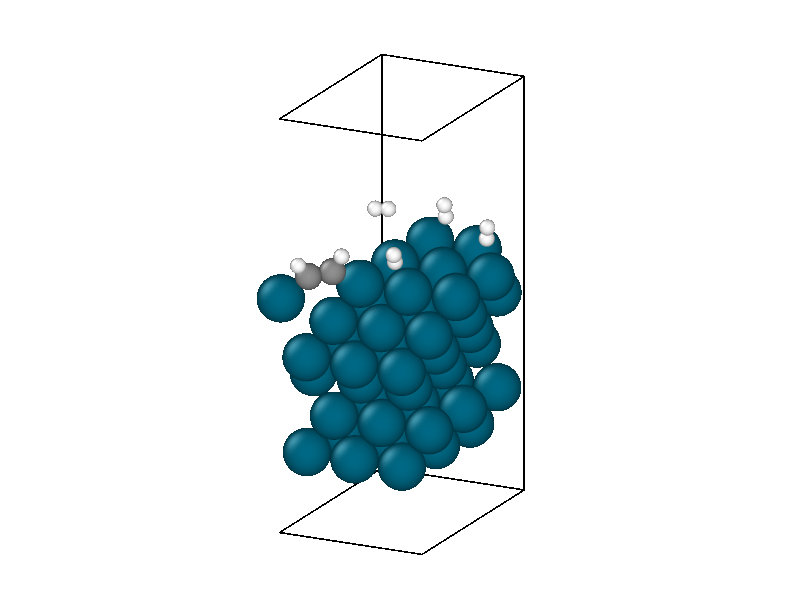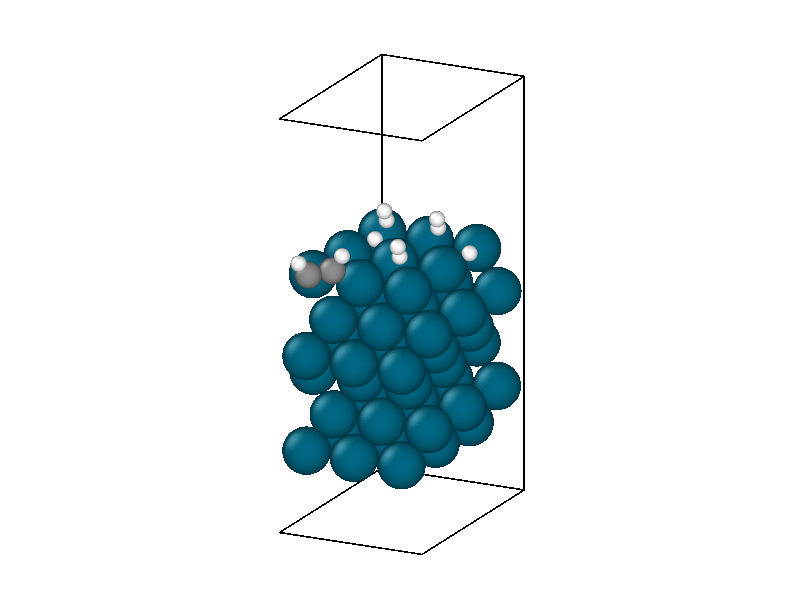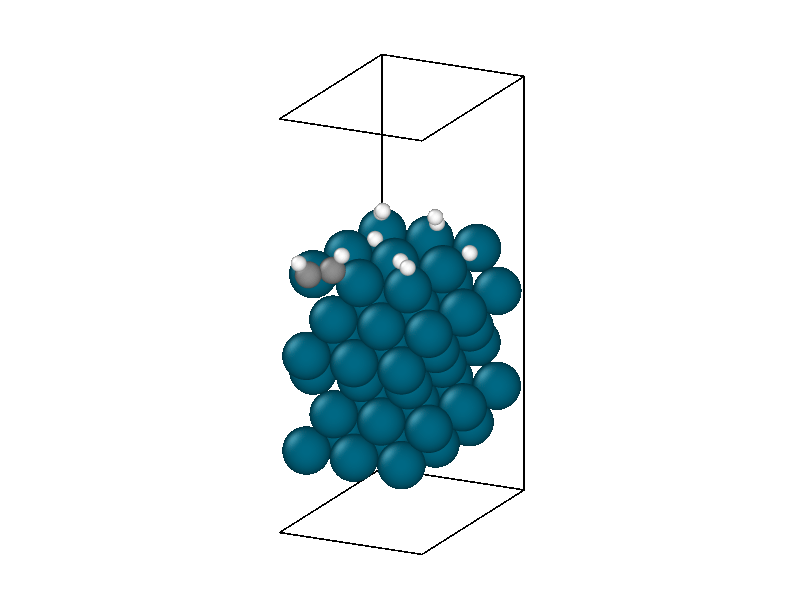
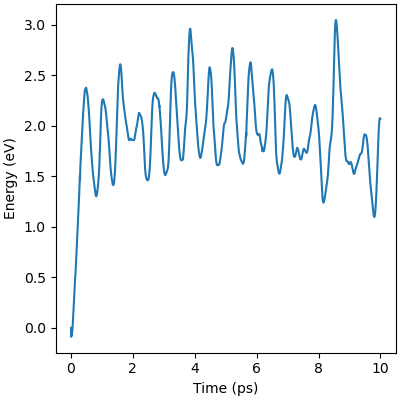
The minimized structure is used to initialize the chemical reaction simulation that consists of a 10ps 300 Kelvin NVT simulation. The NVT simulation recreates the ambient environment of the Horiuti–Polanyi process and provides thermal energy for the remaining hydrogen atoms to decompose and hydrogenate the acetylene. The structural rearrangements are shown in Figure 6. The first phase of the simulation sees the second hydrogen decompose to create four protons. Afterwards, neither of the two remaining hydrogen molecules decomposes, signifying that the active sites are all occupied, four by protons and the other by the adsorbed acetylene.

The thermodynamics are dominated by vibrations in the palladium catalyst masking much of the chemistry. The underlying trend peaks around 5ps and then decreases. This corresponds to the equilibration of the thermal energy released by the second hydrogen decomposing and the protons settling into the hollow sites. After 5ps the hydrogens in the hollow sites begin diffusing into lower energy positions reducing the energy of the system. The hydrogen surface diffusion marks the preliminary step of the Horiuti-Polanyi to move hydrogen atoms to the adsorbed acetylene molecule.
The successful simulation of the Horiuti–Polanyi mechanism for acetylene hydrogenation on a palladium catalyst using PHIN-OS establishes a critical benchmark for future materials development. By accurately reproducing the barrierless hydrogen decomposition and capturing the complex intermediate states of the hydrogenation reaction, this work validates the application of PHIN-OS for complex catalytic processes. This validation moves beyond simply confirming existing experimental observations; it creates a robust, reproducible digital baseline for computational catalysis.
The ultimate significance of this work lies in its potential to accelerate the development of next-generation, PGM-free catalysts. With the validation of the current digital workflow, researchers can confidently substitute alternative, non-precious metal materials into the established PHIN-OS workflow. This platform allows for rapid virtual screening of potential catalyst compositions and surface modifications, enabling the prediction of catalysts in silico before time-consuming and expensive laboratory synthesis. In effect, this research transforms catalyst development from a trial-and-error experimental process into a guided, high-throughput computational search, paving the way for sustainable and cost-effective solutions in industrial hydrogenation processes.